
E.M. Adkins, J.A. Giaccai, J.H. Miller, Computed electronic structure of polynuclear aromatic hydrocarbon agglomerates, Proceedings of the Combustion Institute. 36 (2017) 957–964. doi:10.1016/j.proci.2016.06.186.
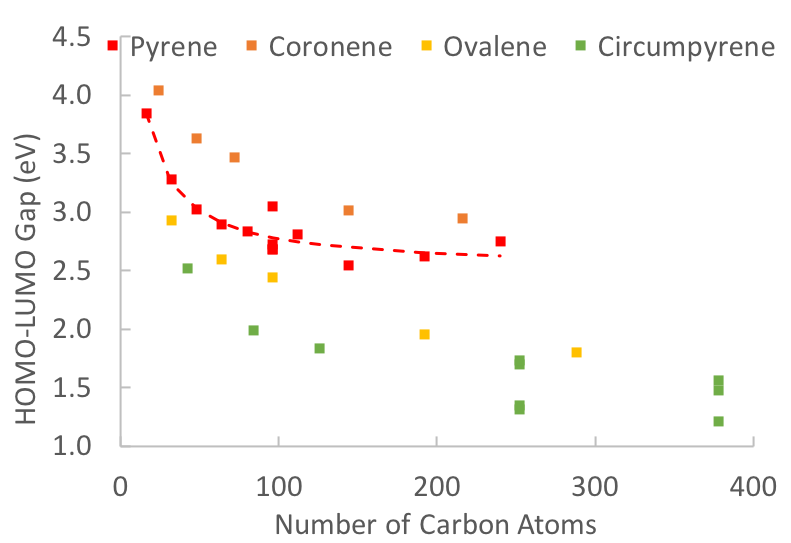
A technique for linking the physical composition of polynuclear aromatic hydrocarbon (PAH) stacks and clusters to their electronic properties is reported. Kohn–Sham HOMO–LUMO gaps are reported for a series of monomers, stacks, and clusters of six, high-symmetry PAHs (pyrene, coronene, ovalene, circumpyrene, circumcoronene, and circumovalene) generated by DFT calculations with the 6-31G* basis set and a B3LYP exchange correlation functional in NWChem. A previously published, atom-pair minimization algorithm was used to optimize the geometries of the PAH stacks and clusters. HOMO–LUMO gaps decrease with an increase in monomer size; homogeneous stacks and clusters indicate substantial lowering of the HOMO–LUMO gap because of agglomeration effects with the formation of dimers and formation of clusters (two or more stacks) being the most dominant contributions. Heteromolecular particulates had HOMO–LUMO gaps that were strongly influenced by the larger components in the system. The HOMO–LUMO gaps of homogeneous clusters approached a maximum agglomeration effect because of the localization of electronic interactions among adjacent stacks. Previously published, experimentally determined optical band gaps (OBG) from Tauc/Davis–Mott analysis of extinction spectra in various laminar, non-premixed flames had an average OBG of 2.1 eV. Based on the computations presented here, this work suggests that clusters with this OBG are comprised of modest molecular size PAH, about the size of ovalene.