
Phys. Chem. Chem. Phys., 2017,19, 28458-28469
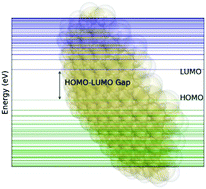 Trends linking the topological characteristics of polynuclear aromatic hydrocarbons (PAH) to their electronic properties are reported. TD-DFT electronic spectra computations, using the 6-31G* basis set and B3LYP exchange correlation functional, were calculated for a series of PAH, allowing for the HOMO–LUMO gaps to be reported. Clar structures provide an avenue to link the physical structure and the aromaticity of the molecule; which, when extended by bond length and harmonic oscillator model of aromaticity analysis, provide powerful tools to understand the link between electronic and physical structure. These results lead to the conclusion that all PAH structures show a decrease in HOMO–LUMO gap as a function of size, but the rate of that decrease is directly related to the topology of the molecules. A PAH taxonomy was developed that categorizes PAH into categories with similar topological properties, which allows for modelling of changes in the HOMO–LUMO gap with PAH size. An atom-pair minimization algorithm was used to calculate the binding energy (BE) of homogeneous dimers of the studied PAH. The BE per carbon atom increases with the overall size of the structure to an asymptotic limit, but as with the HOMO–LUMO gap, topology plays a critical secondary factor. Previously published, experimentally determined optical band gaps (OBG) from Tauc/Davis–Mott analysis of extinction spectra in various laminar, non-premixed flames produced a correlation between the HOMO–LUMO gaps of high-symmetry, nearly circular D2h symmetry molecules to molecular size. The work presented here provides a much more nuanced and predictive evaluation of how OBG depends on structure and size.
Trends linking the topological characteristics of polynuclear aromatic hydrocarbons (PAH) to their electronic properties are reported. TD-DFT electronic spectra computations, using the 6-31G* basis set and B3LYP exchange correlation functional, were calculated for a series of PAH, allowing for the HOMO–LUMO gaps to be reported. Clar structures provide an avenue to link the physical structure and the aromaticity of the molecule; which, when extended by bond length and harmonic oscillator model of aromaticity analysis, provide powerful tools to understand the link between electronic and physical structure. These results lead to the conclusion that all PAH structures show a decrease in HOMO–LUMO gap as a function of size, but the rate of that decrease is directly related to the topology of the molecules. A PAH taxonomy was developed that categorizes PAH into categories with similar topological properties, which allows for modelling of changes in the HOMO–LUMO gap with PAH size. An atom-pair minimization algorithm was used to calculate the binding energy (BE) of homogeneous dimers of the studied PAH. The BE per carbon atom increases with the overall size of the structure to an asymptotic limit, but as with the HOMO–LUMO gap, topology plays a critical secondary factor. Previously published, experimentally determined optical band gaps (OBG) from Tauc/Davis–Mott analysis of extinction spectra in various laminar, non-premixed flames produced a correlation between the HOMO–LUMO gaps of high-symmetry, nearly circular D2h symmetry molecules to molecular size. The work presented here provides a much more nuanced and predictive evaluation of how OBG depends on structure and size.